Lung Fibrosis
A Breath of Hope Through Research !
Idiopathic pulmonary fibrosis (IPF) is a rare, irreversible disease of unknown cause. It is a chronic disease characterized by progressive scarring of the lungs, leading to an accumulation of collagen-producing cells responsible for stiffening the lungs. As a result, the passage of oxygen and the rejection of carbon dioxide are no longer ensured, and patients suffer mainly from significant respiratory difficulties such as progressive breathlessness, both on exertion and at rest, a chronic dry cough and extreme fatigue. IPF generally begins after the age of 50 and has a fairly poor prognosis, with a median survival of around 5 years after diagnosis. At least 12,000 people are thought to be affected in France, with at least 4,400 new cases each year. Treatments remain very limited, with 2 compounds, pirfenidone and nintedanib, showing efficacy in reducing disease progression without completely halting it. Diagnostic difficulties and the lack of tools for monitoring disease progression and/or treatment efficacy are major clinical challenges. Our aim is to find nuclear imaging tools that will facilitate early diagnosis and monitoring of patients, in order to improve their care and survival.
Our team has obtained ANR funding (HYMAGE-IPF) of 240,000 euros over 3 years (2020-2023) to develop, in collaboration with the INSERM HSP-pathies team headed by Dr Carmen Garrido and the CHU de Dijon rare lung disease reference center headed by Pr Philippe Bonniaud, imaging tracers for IPF targeting :
HYPOXIA
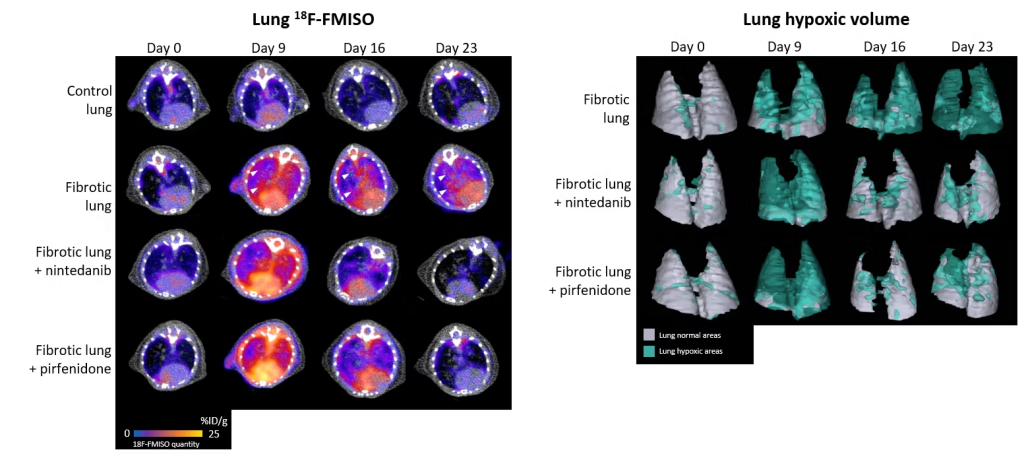
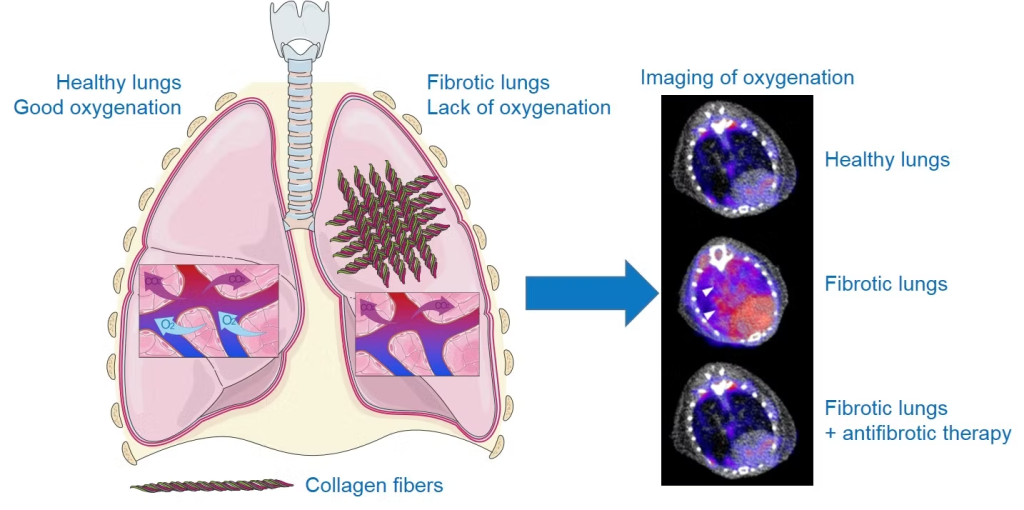
Due to the destruction of lung structure by excess of scar tissue, which disrupts gas exchange, hypoxia (lack of oxygen) is an important feature of the lungs of patients with IPF. Thanks to the use of a radiotracer specific to hypoxic regions, 18F-FMISO (a radioactive molecule that accumulates specifically in cells under oxygen-deprived conditions), we were able to show that areas of hypoxia were more prominent in fibrotic lungs, correlating with disease severity. Moreover, the level of pulmonary hypoxia measured by 18F-FMISO imaging is a good predictive marker of disease progression and response to the anti-fibrosis treatments nintedanib and pirfenidone in our preclinical models (for more details, see the associated publication).
Associated publications:
Bellaye PS, Beltramo G, Burgy O, Collin B, Cochet A, Bonniaud P. Measurement of hypoxia in the lung in idiopathic pulmonary fibrosis: a matter of control. Eur Respir J. 2022 Jan 27:2102711. doi: 10.1183/13993003.02711-2021.
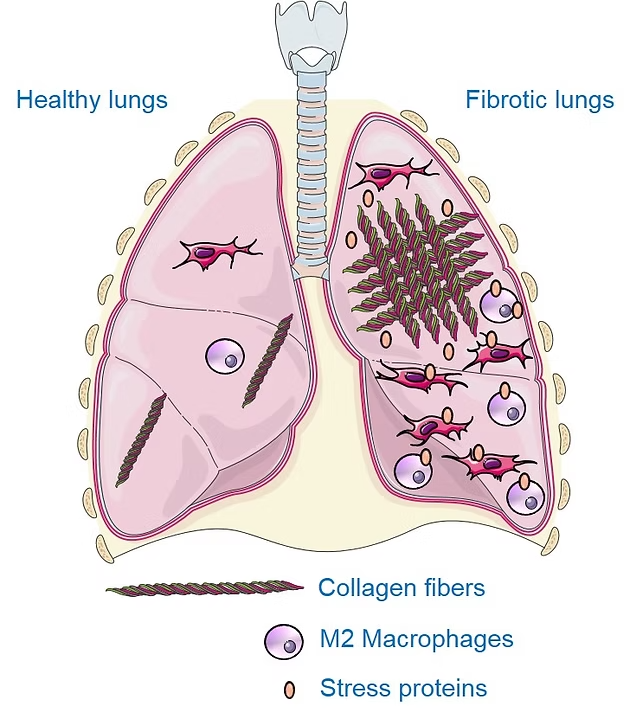
M2 MACROPHAGES
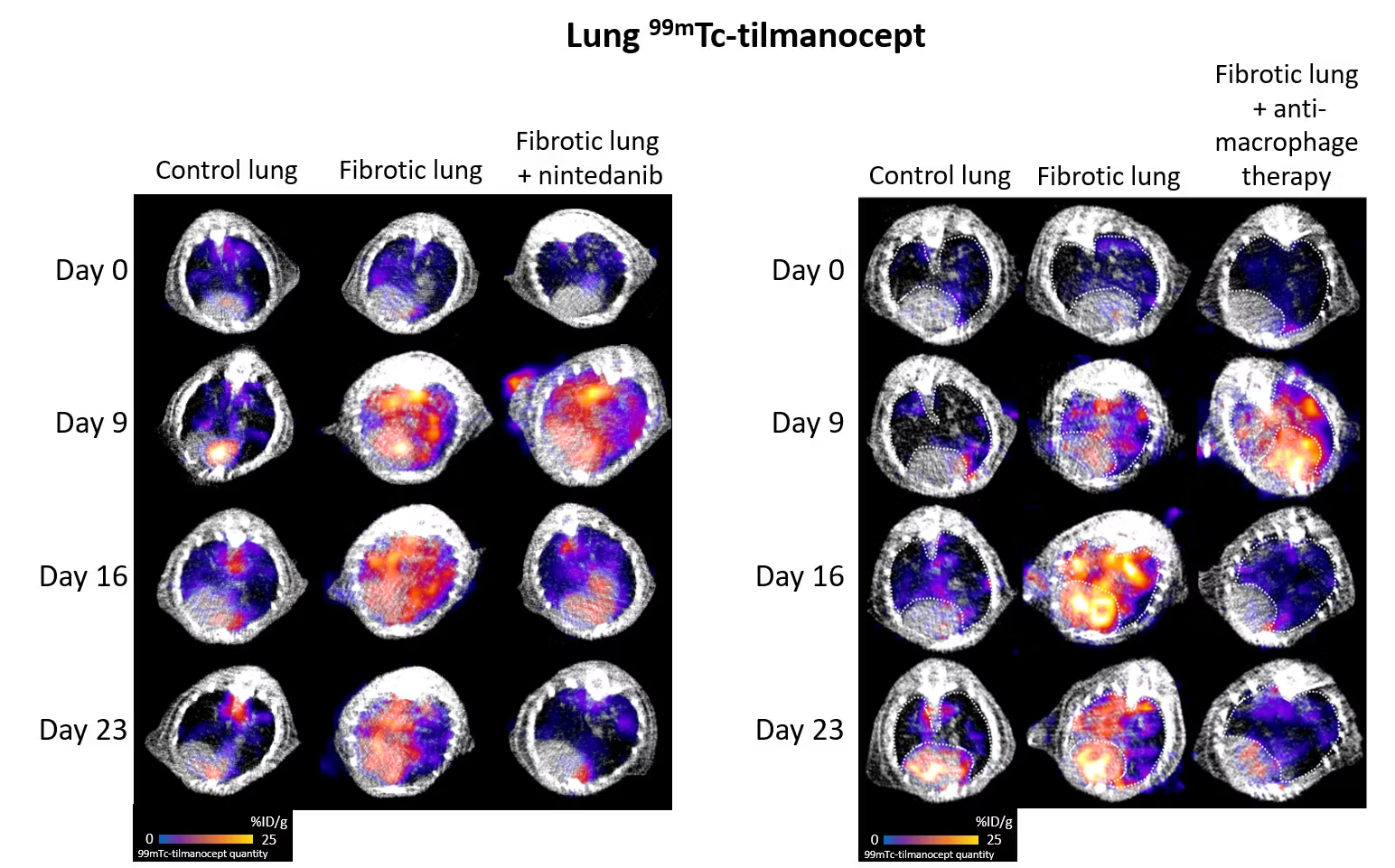
Macrophages are immune cells that circulate and/or reside in various organs, notably the lungs, where they play a part in defending against infection. In the context of fibrosis, these macrophages take on a particular phenotype, known as M2 macrophages, and secrete growth factors, notably TGF-β, which promote the production of scar tissue including collagen. Thanks to the use of a radiotracer specific to M2 macrophages, 99mTc-tilmanocept (a radioactive molecule that binds specifically to M2 macrophages), we were able to show that M2 macrophages were more present in lungs under conditions of fibrosis. In addition, the level of lung M2 macrophages measured by imaging with 99mTc-tilmanocept enables precise monitoring of disease progression and response to the anti-fibrosis treatment nintedanib. Finally, inhibition of these aggressive pro-fibrosis macrophages reduces the progression of pulmonary fibrosis in our preclinical models.
Associated publications:
COLLAGEN
The hallmark of pulmonary fibrosis is an abnormal and massive increase in extracellular matrix (ECM) deposition, mainly collagen, which disrupts the alveolar architecture and may reflect disease progression. In our study, collagen deposition was monitored in bleomycin-induced lung fibrosis in mice by in vivo molecular imaging using a collagen-targeted radiopharmaceutical, 68Ga-collagelin. Fibrosis progression was also monitored using computed tomography, the gold standard technique to detect lung fibrosis in patients.
We demonstrated that the bleomycin-induced increase in collagen lung content can be accurately quantified by 68Ga-collagelin PET imaging in correlation with disease stage and severity. The lung uptake of 68Ga-collagelin was mainly found in fibrotic areas of lungs in bleomycin-receiving mice. Most interestingly, [68Ga]Ga-NODAGA-collagelinPET imaging allowed the in vivo non-invasive monitoring of nintedanib efficacy as well as the anti-fibrotic effect of the JAK inhibitor, tofacitinib.
Thus, collagen-targeted PET imaging appears as a promising non-invasive tool for staging, monitoring and prediction of disease progression and therapy efficacy towards personalized medicine in IPF.
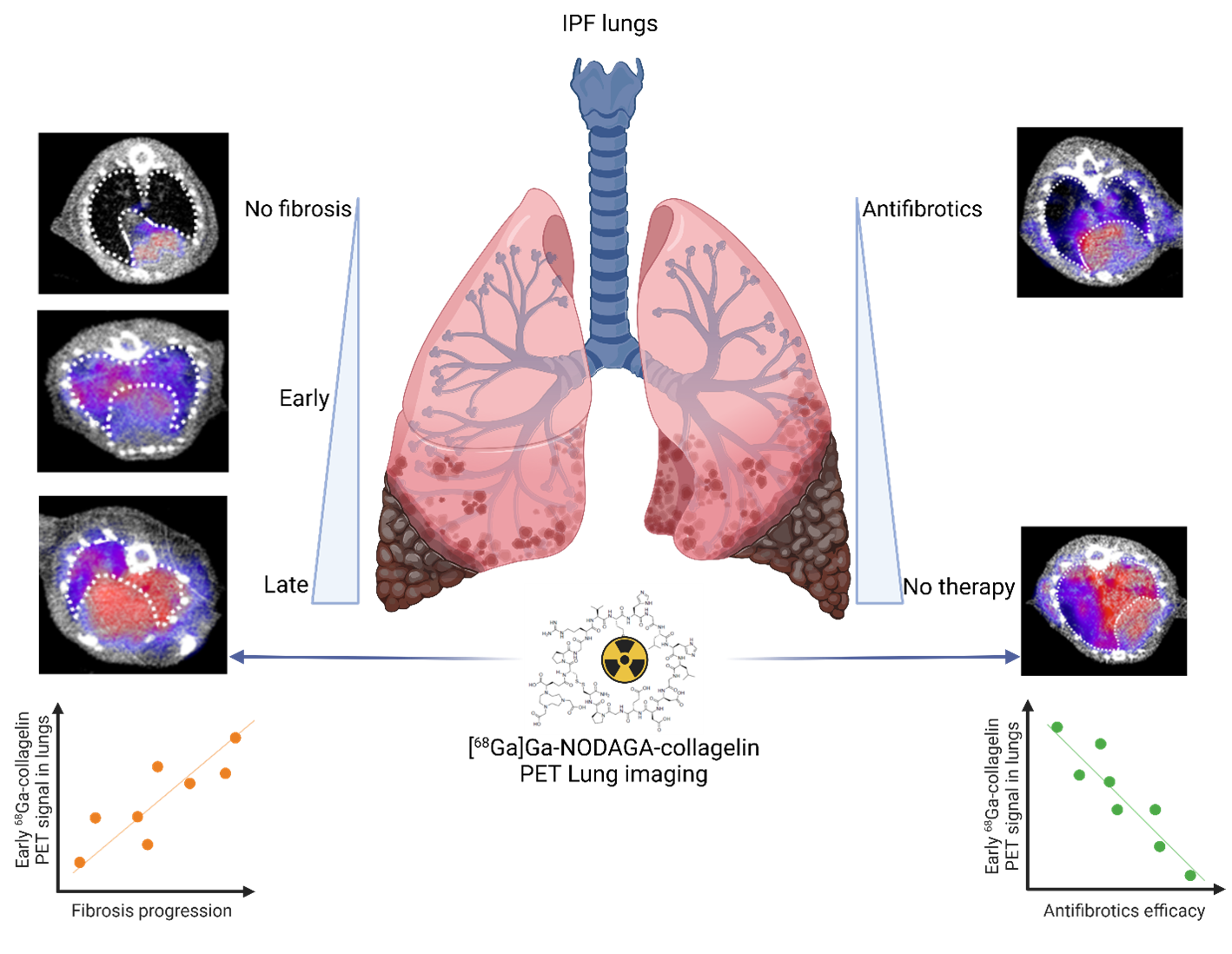
Associated publications:
HEAT SHOCK PROTEINS
Heat shock proteins (or stress proteins, HSPs) are proteins overexpressed under stress conditions to ensure cell survival. In certain conditions, notably pulmonary fibrosis, these proteins promote disease progression. Our team has already shown that HSPs are involved in the overproduction of scar tissue in fibrosis, and that their inhibition is a promising strategy for combating this disease. These are therefore interesting targets for the development of imaging agents to visualize the progression of fibrosis and the response to treatment. Our team is focusing on the HSP90 and Gp96 proteins, which are overexpressed and/or secreted in high quantities in patients with IPF and in preclinical models. Specific radioactive probes for imaging HSP90 and Gp96 in fibrosis are currently being developed at IMATHERa.
Associated publications:
P. S. Bellaye, O. Burgy, P. Bonniaud, M. Kolb, HSP47: a potential target for fibrotic diseases and implications for therapy. Expert Opin Ther Targets 25, 49-62 (2021).